Research overview
Our overall goal is to understand genetic variants that underlie human disease and how their effects vary across different populations. We are a multidisciplinary lab and include both computational and wet lab biologists. We are particularly interested in repetitive DNA variants known as tandem repeats (TRs). Our work is often done in collaboration including with groups in the Depts. of Pediatrics, Medicine, Biomedical Informatics, Computer Science & Engineering, Electrical and Computer Engineering, Chemistry, and Psychiatry at UCSD as well as at other institutions. We are especially interested in building new collaborations with clinicians. We currently focus on the specific areas described below.

|
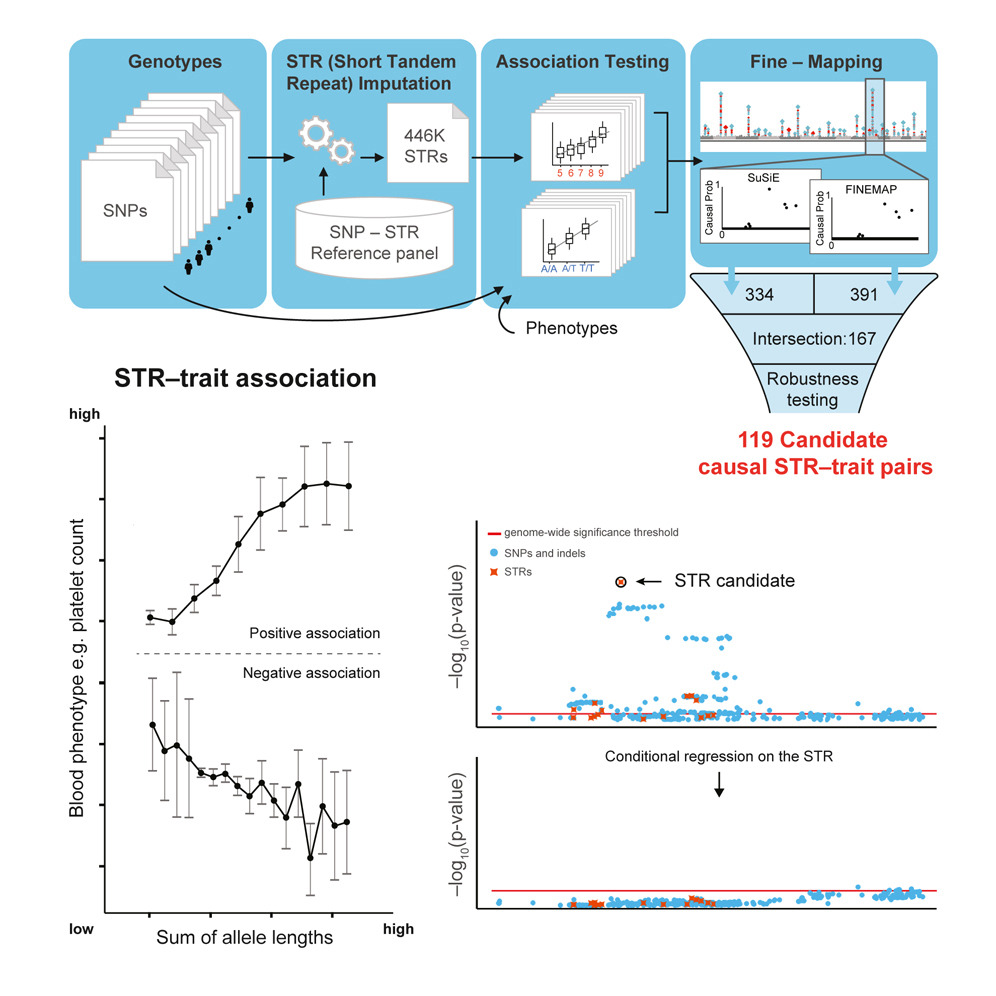
(1) Developing computational tools for analyzing complex variation in biobank-scale genomic datasetsTandem repeats (TRs) are some of the most polymorphic regions of the genome and make outsized contributions to disease but are technically challenging to analyze. Our lab develops methods to enable genome-wide analysis of TRs including:
Many of these tools have been applied to large datasets to gain new insights into patterns of TR variation and the contribution of TRs to different traits. Most of these tools including general utilities for filtering, QC, etc. of TR genotype data are packaged in our TRTools package. TR genotypes generated by many studies we've been involved in are available on WebSTR, a site we built in collaboration with the Anisimova Lab at ZHAW. We are also interested in using pangenomes to understand genetic variation at repeats and other complex regions of the genome. Our lab is a member of the Human Pangenome Reference Consortium. We are working on an interactive browser to visualize pangenome data. |
|
(2) Identifying TRs contributing to human traitsWe have developed and applied methods to integrate TRs into association testing frameworks. We have applied these to uncover widespread contributions of TRs to a range of traits including:
We have also developed and applied methods to study de novo mutations at STRs, which we identified as contributing to risk for autism spectrum disorders (Mitra et al. 2021). We are continuing to apply our association testing and de novo analysis frameworks to study the contribution of TRs to other traits including molecular and disease phenotypes. We also have multiple ongoing collaborative projects to perform genome-editing of predicted pathogenic TRs in human iPSCs and other cell types. |

|
(3) Studying mutation and selection processes at TRs within and across speciesTandem repeats have multiple interesting properties compared to other types of variation, including rapid mutation rates and high rates of multi-allelicness. Understanding the evolutionary forces including mutation and selection driving patterns of variation at these loci is critical to predicting which TR mutations are likely to be pathogenic. We have made multiple contributions including:
|
|
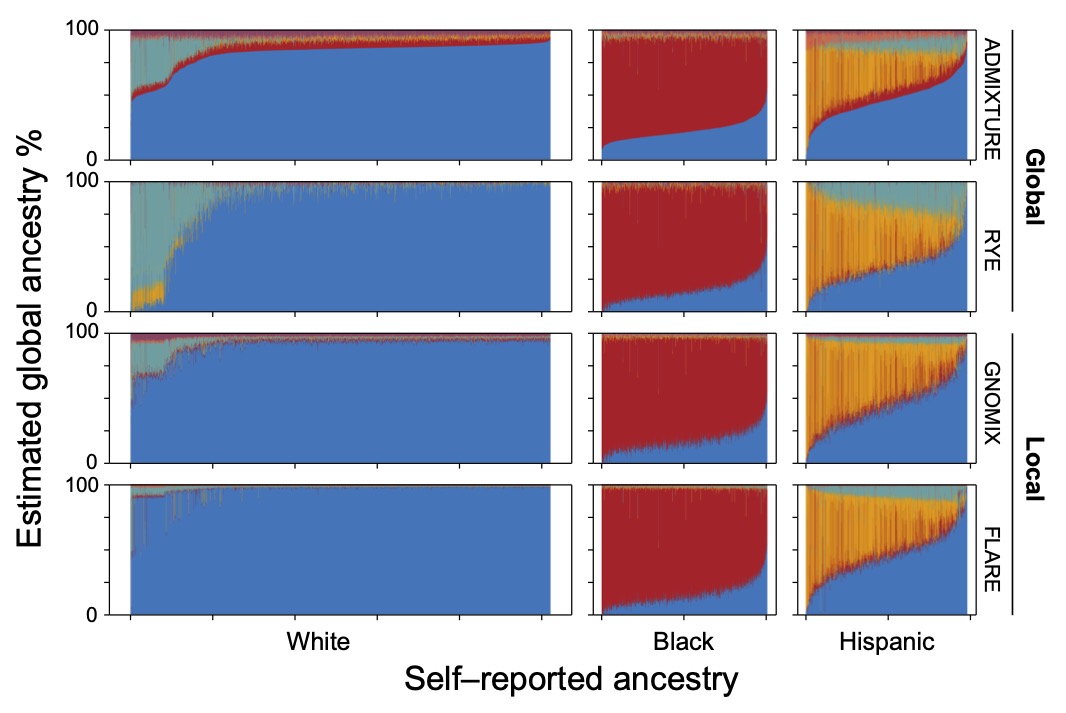
(4) Understanding how the effects genetic variants differ across human populationsTogether with Drs. Kelly Frazer (UCSD) and Lucila Ohno-Machado (Yale), we lead the Center for Admixture Science and Technology (CAST), which focuses on improving the utility of genomics methods for admixed populations. Through CAST we work on multiple projects including:
|
|
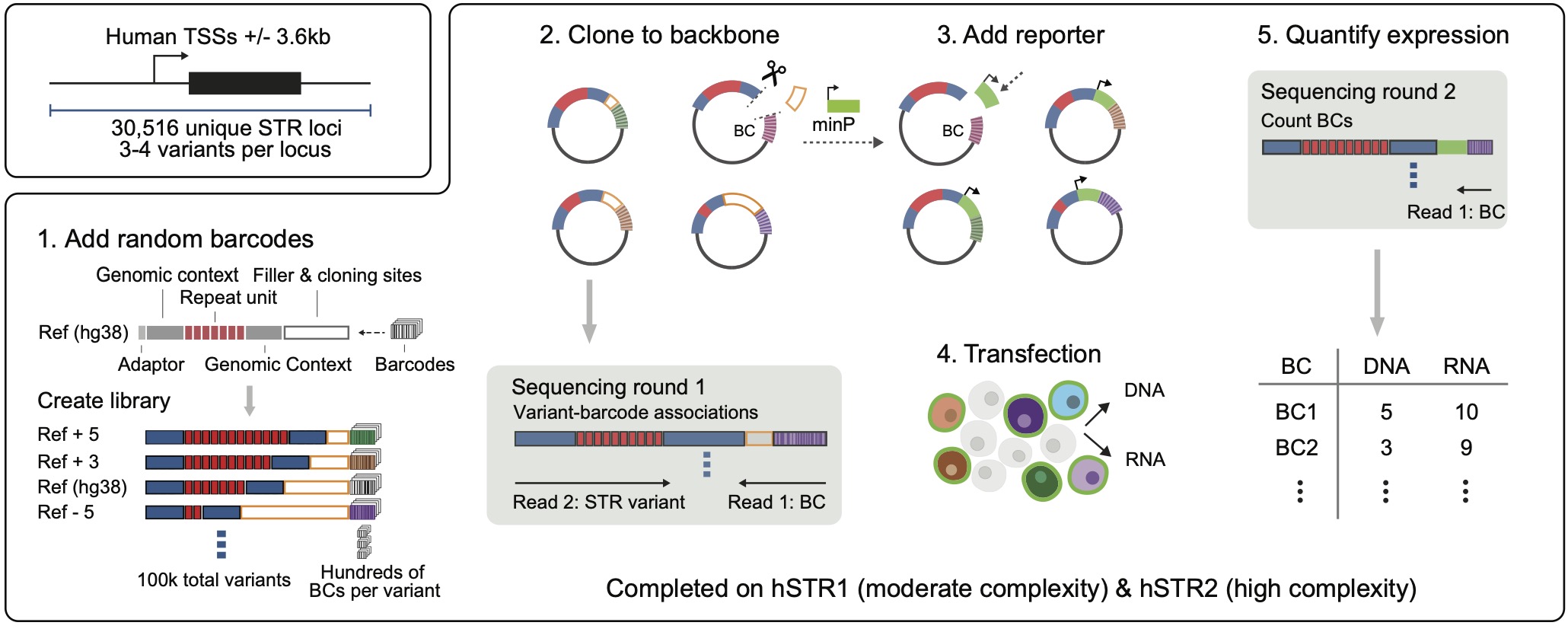
(5) Using high-throughput experimental techniques to study the impacts of genetic variants on molecular and cellcular phenotypesWe have multiple collaborative projects to study the impact of genetic variants in human cells including:
|